
When a drug goes straight into your bloodstream - through an IV, injection, or infusion - there’s no second chance. Your body doesn’t get to filter it out like it does with pills or liquids you swallow. That’s why sterile manufacturing for injectables isn’t just about cleanliness. It’s about precision, control, and absolute certainty that every single dose is free from harmful microbes, particles, or toxins. One contaminated vial can kill. And history has proven it.
Back in 1955, over 200 children died after receiving a polio vaccine that wasn’t properly sterilized. That disaster forced regulators to create the first real rules for sterile drug production. Today, those rules are stricter than ever. The goal? A contamination rate of less than one in a million. That’s the sterility assurance level (SAL 10^-6) set by the WHO. It sounds impossible, but in sterile manufacturing, it’s the bare minimum.
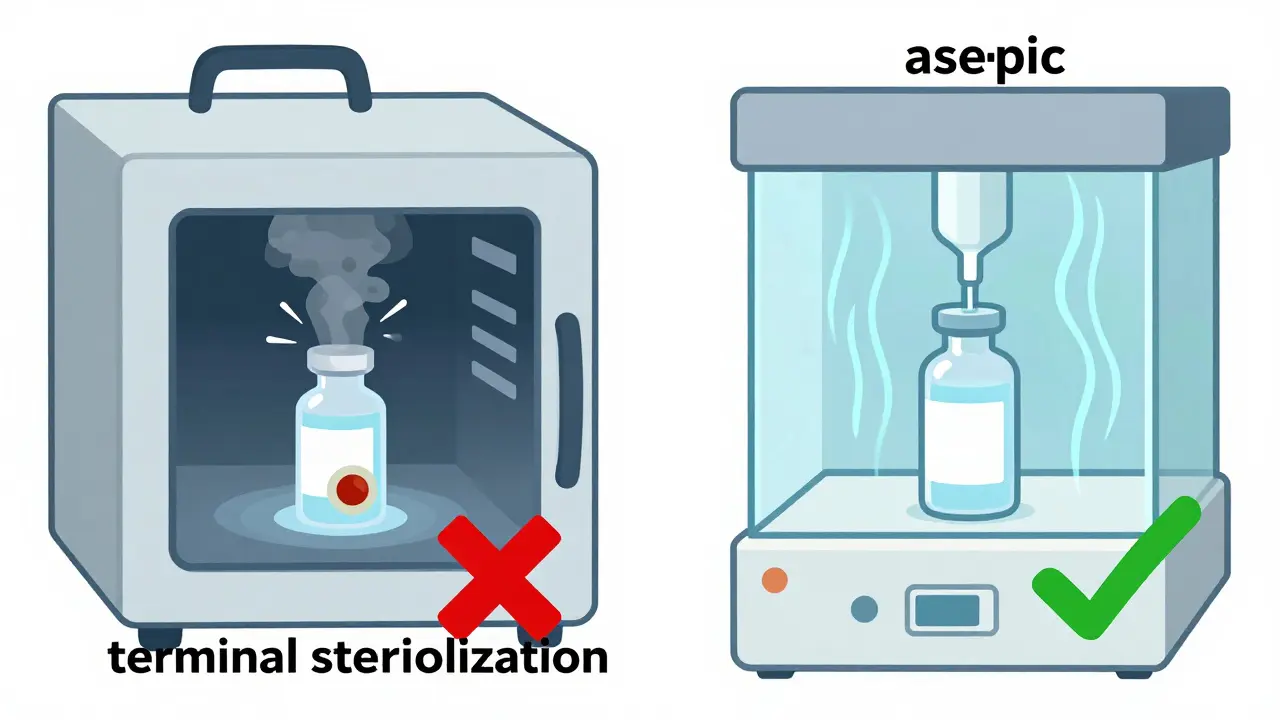
Two Paths to Sterility: Terminal vs. Aseptic
There are two main ways to make injectables sterile. One is called terminal sterilization. The other is aseptic processing. They’re not interchangeable. Choosing the wrong one can ruin a drug - or worse, kill someone.
Terminal sterilization means you make the product, seal it in its final container, then blast it with steam at 121°C for 15 to 20 minutes. Or you zap it with gamma radiation. This kills everything. It’s simple, reliable, and gives you a sterility level of 10^-12 - better than the required 10^-6. But here’s the catch: heat and radiation destroy many modern drugs. If your medicine is a protein, antibody, or gene therapy, it’ll break down. That’s why only 30-40% of injectables can use this method, according to Vetter Pharma’s 2022 report.
The other method - aseptic processing - is where most new drugs are made. No heat. No radiation. Instead, everything happens in a cleanroom so controlled, it’s like working inside a spaceship. Workers wear full-body suits. Air is filtered through HEPA filters. Every surface is wiped down. The filling area itself must meet ISO 5 standards: fewer than 3,520 particles per cubic meter that are 0.5 microns or larger. That’s smaller than a bacterium. And even then, you’re not done.
What Makes a Cleanroom Work
A cleanroom isn’t just a room with fancy filters. It’s a system. Pressure matters. Airflow matters. Temperature and humidity matter. If the pressure between rooms isn’t tight - say, 10-15 Pascals higher in the ISO 5 zone than the surrounding ISO 7 zone - contaminants will sneak in. If the air doesn’t turn over 20 to 60 times per hour, particles settle. If humidity drops below 45% or rises above 55%, static electricity builds up, pulling dust into the product.
And then there’s the water. Not just any water. It has to be Water for Injection (WFI). That means it’s distilled, filtered, and tested to have less than 0.25 endotoxin units per milliliter. Endotoxins are toxins from dead bacteria. Even if you kill all the bugs, their leftover poison can still cause fever, shock, or death. That’s why containers are baked at 250°C for 30 minutes - to burn off any lingering endotoxins. That’s called depyrogenation. Skip this step, and your drug could be lethal.
Technology That Makes the Difference
For aseptic filling, you need either RABS (Restricted Access Barrier Systems) or isolators. RABS are like sealed gloveboxes with transparent walls. Technicians reach in through gloves to fill vials. Isolators are fully enclosed, automated chambers. No human contact at all. Dr. James Akers from the BioPharmaceutical Technology Center says isolators reduce contamination risk by 100 to 1,000 times compared to older cleanrooms. But they cost 40% more to install.
That’s why some companies stick with RABS. The Parenteral Drug Association says if you train your staff well and maintain your gloves, RABS can perform just as well. But one glove tear - and you’ve lost your batch. In October 2023, a major pharma company lost $450,000 in one day because of a tiny tear in a RABS glove. They didn’t even know until the media fill test came back positive.
Media fill tests are mandatory. You simulate the whole process using growth media instead of real drug. Then you incubate it. If even one vial grows bacteria? Your entire process is flawed. The FDA says if more than 0.1% of your media fills fail, your system isn’t under control. That’s one bad vial out of every thousand. And if that happens, you shut down.
Costs, Risks, and Real-World Failures
Sterile manufacturing is expensive. Terminal sterilization runs about $50,000 per batch. Aseptic processing? $120,000 to $150,000. Why? Because you need more space, more filters, more monitoring, more training. Every hour of operation costs more. But the real cost isn’t the money - it’s the risk.
In 2012, the New England Compounding Center shipped 17,000 contaminated steroid injections. 751 people got sick. 64 died. The cause? A dirty room. Untrained staff. No real-time air monitoring. The FDA’s 2022 inspection data shows 68% of sterile manufacturing violations are about aseptic technique - not equipment failure. People mess up. Gloves get torn. Hands brush against surfaces. Training isn’t enough. You need constant oversight.
A survey of 45 sterile facilities found 68% had at least one sterility failure per year. Each one cost an average of $1.2 million. That’s not just lost product. It’s delayed treatments, lost trust, lawsuits. One company reduced defects from 0.2% to 0.05% by switching to automated visual inspection. But it cost $2.5 million. Was it worth it? Absolutely. Because one patient death is one too many.

What’s Changing Now
Regulations are tightening. The EU’s revised Annex 1 in 2022 requires continuous air and particle monitoring - not just spot checks. The FDA’s 2023 guidance pushes for real-time data, closed systems, and fewer manual interventions. More companies are switching to closed processing. In 2023, 65% of new sterile facilities used them. That’s up from 30% in 2019.
Robots are taking over filling lines. Rapid microbiological tests are cutting wait times from 14 days to 24 hours. Digital twins - virtual copies of your production line - let you test failures before they happen. The FDA’s 2024-2026 plan even includes AI tools to predict contamination risks before inspections.
But the biggest shift? Who’s making these drugs. Contract manufacturers now produce 55% of sterile injectables. Companies like Lonza, Catalent, and Thermo Fisher handle most of the work. That means your drug’s safety isn’t just your responsibility - it’s theirs. And if they cut corners? You’re on the hook.
Why This Matters to You
If you’re taking an injectable - whether it’s insulin, chemotherapy, or a new biologic - you’re relying on dozens of people working in a room where the air is cleaner than a hospital operating theater. Every step, from the water used to the gloves worn, is designed to keep you safe. But safety isn’t automatic. It’s engineered. It’s tested. It’s monitored. And if any one part fails, the consequences are deadly.
The global market for sterile injectables hit $225 billion in 2023. It’s growing fast. More than 40% of new drugs need this kind of manufacturing. But as demand rises, so does pressure to cut costs. That’s why the FDA’s inspection citations jumped from 1,245 in 2019 to 1,872 in 2022. Companies are racing to produce more - but not always safely.
There’s no room for shortcuts. Not here. Not ever.
What’s the difference between terminal sterilization and aseptic processing?
Terminal sterilization kills microbes after the product is sealed, using heat or radiation. It’s reliable but only works for drugs that can survive high temperatures. Aseptic processing keeps everything sterile during manufacturing without using heat. It’s used for sensitive drugs like biologics but requires extreme control over air, equipment, and personnel.
Why is Water for Injection (WFI) so important?
WFI is the only water approved for injectables because it’s purified to remove not just microbes, but also endotoxins - toxic substances left behind by dead bacteria. Even if you sterilize the drug, endotoxins in the water can cause fever, shock, or death. WFI must have less than 0.25 EU/mL of endotoxins, as required by USP <85>.
What is a media fill test?
A media fill test simulates the entire aseptic manufacturing process using nutrient-rich liquid instead of the actual drug. After filling, the vials are incubated to see if any bacteria grow. If even one vial shows contamination, the entire process is flawed and must be fixed before production resumes. It’s the gold standard for proving your sterile process works.
Why are isolators better than RABS?
Isolators are fully enclosed, automated systems that eliminate human contact with the product, reducing contamination risk by 100 to 1,000 times compared to RABS. RABS rely on operators reaching in through gloves, which introduces risk. While properly operated RABS can match isolator performance, isolators offer more consistent control - especially for high-risk products.
What happens if a sterile manufacturing facility fails an FDA inspection?
If a facility fails, the FDA can issue a Form 483 listing violations. If serious - like repeated media fill failures or poor environmental monitoring - the agency may halt production. Products already shipped may be recalled. The company can’t sell new batches until it fixes the issues and passes a re-inspection. In extreme cases, the facility may be banned from importing drugs into the U.S.
How often do sterility failures happen in real-world facilities?
A 2022 BioPhorum survey of 45 sterile manufacturing facilities found that 68% had at least one sterility failure per year. The average cost per failure was $1.2 million. Most failures trace back to human error, glove breaches, or inadequate environmental monitoring - not equipment breakdowns.
Are sterile injectables safer now than they were 10 years ago?
Yes - but not because of luck. Advances in technology like continuous monitoring, closed systems, automation, and digital twins have reduced contamination risks significantly. However, regulatory scrutiny has also increased. Facilities that haven’t upgraded their systems since 2020 are now at higher risk of failure. Safety has improved, but only for those who invest in modernization.



Mary Beth Brook
March 8, 2026 AT 12:05Sterility Assurance Level 10^-6 isn't a goal-it's a floor. If you're hitting that and calling it a day, you're already behind. Terminal sterilization isn't obsolete-it's the gold standard. Anything else is just playing Russian roulette with a syringe.
WHO doesn't set rules. They enforce them. And if your facility can't meet SAL 10^-6, you shouldn't be touching a vial.
Stop romanticizing aseptic. It's a high-risk workaround. Not a breakthrough.
Neeti Rustagi
March 8, 2026 AT 13:22Dear colleagues, it is with profound respect for public health that I must emphasize the gravity of aseptic technique failures.
Water for Injection is not merely purified-it is sanctified. Endotoxin levels below 0.25 EU/mL are not a suggestion; they are a covenant with life itself.
When we compromise on environmental controls, we do not merely risk contamination-we betray the trust of the patient who trusts their life to our precision.
Let us not forget: every vial carries not just medication, but hope. And hope, once broken, cannot be recalled.
Dan Mayer
March 8, 2026 AT 19:48ok so i read this whole thing and like... why are we still using HEPA filters? like seriously? they're 1960s tech. we got nanofibers now. and why are we even doing media fills? why not just do real-time PCR on every batch? like duh.
also the 1955 polio thing? that was because they used monkey kidney cells. we don't do that anymore. so why are we still using that as a scare tactic? lmao.
also rabs? please. isolators are the only way. unless you're a masochist. or a budget CFO. then i guess you're just asking for a lawsuit.
Janelle Pearl
March 9, 2026 AT 01:00I’ve worked in three different sterile facilities. I’ve seen gloves tear. I’ve seen a tech sneeze into a laminar flow hood. I’ve watched someone forget to change their booties.
And you know what? We didn’t lose a single batch. Not because we were perfect. But because we had layers. Redundancy. Oversight. Culture.
It’s not about the tech. It’s about the people who show up, every shift, and choose to care.
That’s what keeps people alive. Not the isolators. Not the audits. The people.
Ray Foret Jr.
March 10, 2026 AT 11:04Man, this post hit different. 😔
I used to work in pharma QA and we had a media fill fail because someone left a pen cap inside the RABS. Just... a cap.
Cost us $800k. Shut down for 11 days.
But you know what? We fixed it. We trained harder. We made videos. We laughed about it. And now? Our failure rate is 0.01%.
It’s not perfect. But we’re getting there. 🙌
One vial at a time.
Samantha Fierro
March 11, 2026 AT 07:39The regulatory landscape has evolved dramatically since Annex 1 and the FDA’s 2023 guidance. Continuous environmental monitoring is no longer optional-it is foundational.
Furthermore, the shift toward closed-system processing reflects a mature understanding of human factor risk. Automation is not a luxury; it is an ethical imperative.
Facilities that cling to legacy RABS without rigorous glove integrity testing are operating with unacceptable risk profiles.
Investment in digital twins and AI-driven predictive analytics is not merely prudent-it is a moral obligation to patient safety.
Robert Bliss
March 12, 2026 AT 10:03Honestly? I don't need all the jargon. All I know is: if someone's getting an IV, they're trusting that no one messed up.
It's not about robots or isolators. It's about someone caring enough to check the glove, clean the surface, and double-check the water.
Simple stuff. But it saves lives.
Let's not forget that.
Thanks for writing this. Needed to hear it.
Peter Kovac
March 13, 2026 AT 08:37Let’s be clear: the 68% failure rate cited is misleading. It aggregates facilities with decades-old infrastructure alongside those with modern closed systems.
Furthermore, the $1.2 million average cost per failure is a red herring. It ignores the opportunity cost of delayed therapies and the reputational damage to the entire industry.
Most importantly, the root cause analysis is absent. Human error is a symptom-not the cause. The real failure is in management’s decision to underfund validation, overwork staff, and ignore real-time data.
There is no ‘accident.’ There is only negligence.
APRIL HARRINGTON
March 14, 2026 AT 01:15OMG I JUST READ THIS AND I CRIED 😭
64 people died from a dirty room???
And we’re still letting people reach into gloveboxes??
And the water? ENDOTOXINS??
Who even knows what that means??
My cousin got chemo last year and I didn’t even think about this
How is this not on the news??
WHY ISN’T THERE A MOVIE ABOUT THIS??
WE NEED TO DO SOMETHING
RIGHT NOW
LIKE TOMORROW
PLEASE
Leon Hallal
March 15, 2026 AT 17:00You think this is bad? Wait till you hear about the 2021 case where a technician used tap water to wipe down a vial. Because he thought ‘WFI’ stood for ‘Water for Injection’… and also ‘Water for Inhaling’.
He got fired. The batch was destroyed. But the real tragedy? He still believes he did nothing wrong.
That’s the real epidemic. Not the microbes. The arrogance.
Judith Manzano
March 16, 2026 AT 00:04This is so fascinating. I’ve never thought about how water is processed for injections. I just assumed it was like bottled water.
But endotoxins? Depyrogenation? SAL 10^-6?
I had to look up all of this. And now I’m terrified. But also… impressed.
How do we get this info out to more people? This isn’t just for scientists. This is for everyone who takes medicine.
Maybe we need a short video. Or a comic. Something simple.
Because if we don’t understand the stakes, we can’t demand better.
rafeq khlo
March 16, 2026 AT 02:09Contract manufacturers are the real problem. They outsource the cleanrooms, outsource the training, outsource the blame.
And then they charge $150k per batch and call it ‘value-added’.
Meanwhile, the brand owner sits in a boardroom sipping bourbon, thinking they’re safe.
They’re not.
They’re just paying someone else to risk their life.
And the FDA? They inspect the brand. Not the contractor.
That’s not oversight. That’s negligence dressed as regulation.
Morgan Dodgen
March 16, 2026 AT 07:34Who really controls the sterile manufacturing space? Not the scientists. Not the regulators.
It’s the same 3 conglomerates that own the filters, the isolators, the validation software.
And they’re the ones who wrote the guidelines.
Remember how the FDA ‘discovered’ the media fill requirement? It was after a lobbying campaign from a company that sold automated fill systems.
They didn’t improve safety.
They created a dependency.
And now? You can’t get certified unless you buy their $2M system.
It’s not about sterility.
It’s about monopoly.
Philip Mattawashish
March 18, 2026 AT 04:01Terminal sterilization is the only honest path. Everything else is theater.
Aseptic processing is a Ponzi scheme built on cleanroom illusions and human fallibility.
You think your ISO 5 zone is safe? You’re deluding yourself. The air is never clean. The gloves are never intact. The training is never enough.
And yet we pretend.
We pretend because admitting the truth would mean shutting down 70% of modern biologics.
So we lie to patients. We lie to regulators. We lie to ourselves.
And we call it science.
It’s not.
It’s a funeral march dressed in lab coats.