Imagine your body is a house. You have pipes that carry water in and drains that let it out. Now imagine those drains get clogged. Water starts backing up, flooding the basement, damaging the walls, and eventually ruining the foundation. That is essentially what happens in Wilson’s disease, a rare genetic condition where copper builds up in your organs instead of being flushed out.
Wilson’s disease is an inherited disorder that prevents the body from getting rid of excess copper. Without treatment, this metal accumulates to toxic levels in the liver, brain, kidneys, and eyes. It sounds scary, but here is the good news: if caught early, Wilson’s disease is completely manageable. People with this condition can live normal, healthy lives thanks to treatments called chelation therapy.
What Is Happening Inside Your Body?
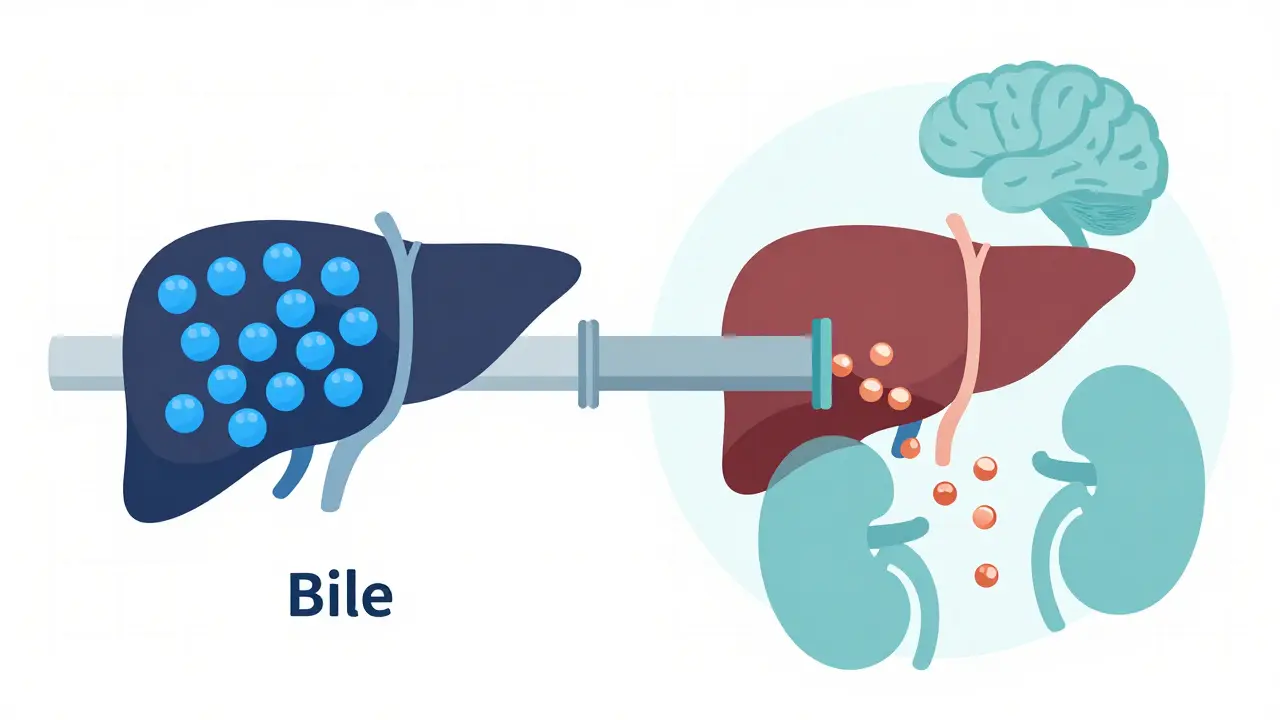
To understand Wilson’s disease, you first need to know how copper works. Copper is essential. Your body uses it to build red blood cells, maintain nerve health, and keep bones strong. In a healthy person, the liver acts as a gatekeeper. It takes in copper from food, attaches most of it to a protein called ceruloplasmin for safe transport, and dumps the extra into bile so it leaves your body through waste.
In Wilson’s disease, that gatekeeper fails. The problem lies in a specific gene called ATP7B. This gene provides instructions for making a protein that moves copper around inside liver cells. When you inherit two faulty copies of this gene (one from each parent), the protein doesn’t work right. Two critical things go wrong:
- Copper isn’t loaded onto ceruloplasmin: This means low levels of ceruloplasmin in your blood, which doctors use as a key diagnostic marker.
- Copper isn’t excreted into bile: Instead of leaving the body, copper stays in the liver cells.
At first, the liver tries to protect itself by binding the extra copper to another protein called metallothionein. But once those binding sites fill up, the copper spills over. It leaks into the bloodstream and travels to other parts of the body, causing damage wherever it settles.
Recognizing the Signs: Liver vs. Brain Symptoms
Wilson’s disease is tricky because it wears many masks. Symptoms usually show up between ages 5 and 35, but they can appear earlier or later. The symptoms depend on where the copper has accumulated the most.
Liver symptoms are often the first sign, especially in children and young adults. They look like common liver issues, which is why misdiagnosis is so common. You might experience:
- Fatigue and weakness
- Loss of appetite
- Sudden jaundice (yellowing of skin and eyes)
- Swelling in the abdomen or legs
- Dark urine
Neurological symptoms tend to appear later, often in teens or adults. These happen when copper damages the basal ganglia, a part of the brain that controls movement. Look out for:
- Tremors or shaking hands
- Difficulty speaking or swallowing
- Muscle stiffness or rigidity
- Poor balance and coordination
- Mood changes, anxiety, or depression
There is also a telltale eye sign. About 95% of people with neurological Wilson’s disease develop Kayser-Fleischer rings. These are brownish-gold deposits of copper in the cornea, visible only with a special slit-lamp exam by an eye doctor. If you see these rings, it’s a major clue pointing toward Wilson’s disease.
How Doctors Diagnose Wilson’s Disease
Because the symptoms mimic other conditions like autoimmune hepatitis or Parkinson’s disease, diagnosing Wilson’s disease can take time. On average, patients wait nearly three years after their first symptoms before getting the correct diagnosis. To speed this up, doctors use a combination of tests:
- Blood tests: They check for low ceruloplasmin levels (usually below 20 mg/dL) and high free copper levels.
- Urine tests: A 24-hour urine collection measures how much copper your body is trying to flush out. In Wilson’s disease, this number is often above 100 micrograms per day.
- Eye exam: An ophthalmologist looks for Kayser-Fleischer rings.
- Liver biopsy: In unclear cases, a small sample of liver tissue is tested for copper content. High levels confirm the diagnosis.
- Genetic testing: Looking for mutations in the ATP7B gene provides a definitive answer.
Doctors often use a scoring system, like the Leipzig criteria, to combine these results. If the score is high enough, treatment begins immediately. Early detection is crucial because liver damage can become irreversible if left untreated.
Chelation Therapy: Removing the Excess Copper
The main goal of treatment is to lower copper levels and prevent further buildup. This is done through chelation therapy. Chelators are drugs that bind to copper in the blood and tissues, forming a complex that your kidneys can filter out in your urine.
There are two primary chelating agents used today:
| Medication | How It Works | Common Side Effects | Best For |
|---|---|---|---|
| D-Penicillamine | Binds copper for urinary excretion | Nausea, metallic taste, rare lupus-like syndrome | Initial treatment due to low cost and availability |
| Trientine | Binds copper for urinary excretion | Fewer side effects than D-penicillamine, higher cost | Patient who cannot tolerate D-penicillamine |
D-Penicillamine has been the standard treatment for decades. It is effective but comes with a catch: about 20-50% of patients experience a temporary worsening of neurological symptoms when they start taking it. This happens because the drug pulls copper out of the brain too quickly. To manage this, doctors often add zinc supplements during the first few weeks.
Trientine is a newer alternative. It tends to cause fewer severe side effects and may be gentler on the nervous system, making it a preferred choice for many patients, especially those with significant neurological involvement. However, it is significantly more expensive.
Zinc Therapy: Blocking Absorption
While chelators remove existing copper, zinc acetate works differently. Zinc doesn’t pull copper out of your body. Instead, it blocks your intestines from absorbing new copper from food.
When you take zinc, it triggers the production of a protein called metallothionein in your gut cells. This protein binds to dietary copper, trapping it in the intestinal lining. When those cells shed and leave the body, the trapped copper goes with them. Zinc is often used for:
- Maintenance therapy: After initial chelation lowers copper levels, zinc keeps them stable.
- Pregnancy: Chelators can cross the placenta and harm the fetus, so zinc is safer for pregnant women.
- Asymptomatic patients: People diagnosed through family screening who haven’t developed symptoms yet.
Zinc is generally well-tolerated, though some people experience mild stomach upset. Taking it on an empty stomach helps absorption, but if nausea occurs, taking it with a light meal is acceptable.
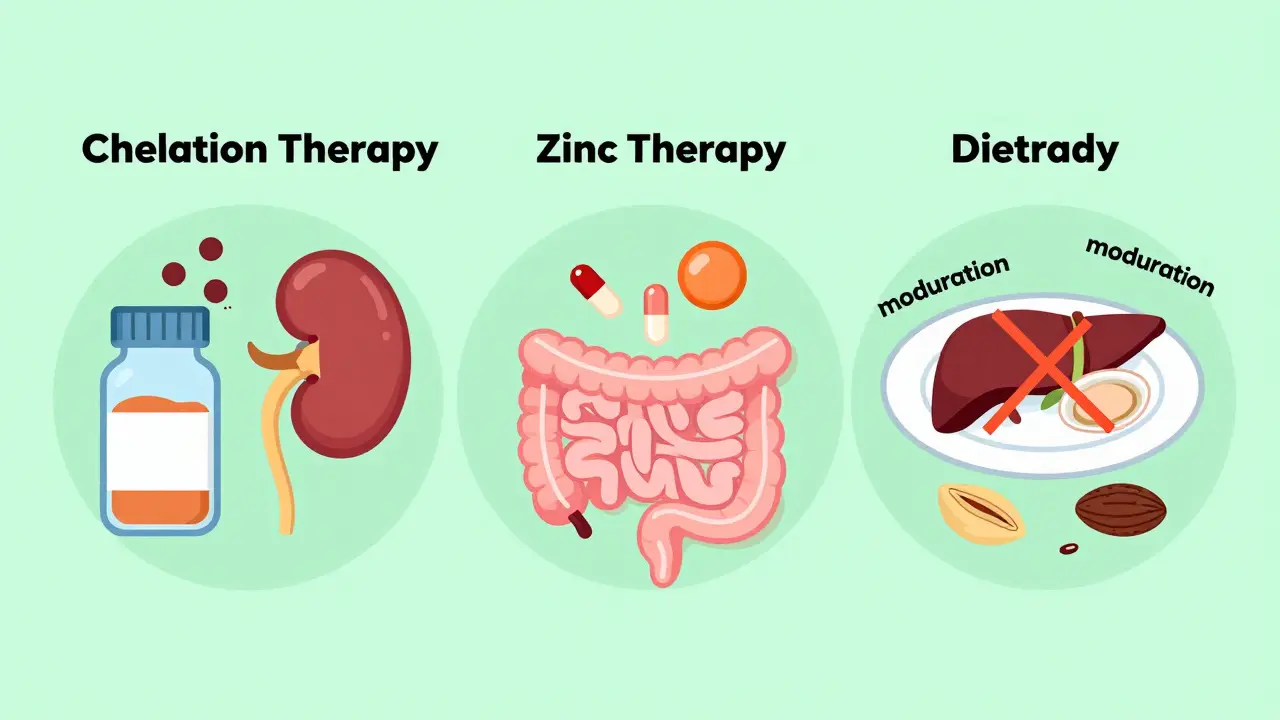
Living with Wilson’s Disease: Diet and Lifestyle
Medication is the cornerstone of treatment, but diet plays a supporting role. You don’t need to eliminate all copper-rich foods, but you should avoid extreme sources. Here are some practical tips:
- Avoid shellfish: Oysters, clams, and mussels are extremely high in copper.
- Limit organ meats: Liver and kidney contain high concentrations of copper.
- Be careful with nuts and seeds: Cashews, sunflower seeds, and chocolate have moderate copper levels. Enjoy them in moderation.
- Check your water: If you have old copper pipes, let the water run for a minute before drinking or cooking to reduce copper exposure.
Regular monitoring is non-negotiable. You will need blood tests every 3 months and urine tests every 6 months to ensure your copper levels stay in the target range. Missing doses can lead to a rapid rebound of copper levels, potentially causing acute liver failure. Setting phone reminders or using pill organizers can help maintain consistency.
Looking Ahead: New Treatments and Hope
Research into Wilson’s disease is advancing rapidly. Scientists are developing new drugs that aim to be more effective with fewer side effects. For example, tetrathiomolybdate is showing promise in clinical trials for its ability to penetrate the blood-brain barrier more effectively than traditional chelators.
Gene therapy is also on the horizon. Early trials are exploring ways to deliver a working copy of the ATP7B gene directly to liver cells, potentially offering a cure rather than just management. While these treatments are still in development, they offer hope for a future where Wilson’s disease requires less intensive daily management.
If you suspect you or a loved one might have Wilson’s disease, don’t wait. Talk to your doctor about testing. With proper care, you can control copper levels and live a full, active life.
Is Wilson’s disease hereditary?
Yes, Wilson’s disease is an autosomal recessive genetic disorder. This means a child must inherit two defective copies of the ATP7B gene (one from each parent) to develop the disease. Parents who carry one copy are usually healthy carriers and do not show symptoms.
Can Wilson’s disease be cured?
There is currently no cure for Wilson’s disease, but it is highly treatable. Lifelong medication is required to manage copper levels. Without treatment, the disease is fatal, but with consistent therapy, patients can have a normal life expectancy.
What are the side effects of chelation therapy?
D-Penicillamine can cause nausea, loss of taste, and rarely, a lupus-like syndrome or kidney problems. Trientine has fewer side effects but may cause iron deficiency. Zinc therapy is generally well-tolerated but can cause mild stomach upset. Regular monitoring helps manage these risks.
How long does it take for symptoms to improve?
Liver enzymes often improve within a few months of starting treatment. Neurological symptoms may take longer, sometimes 6 to 12 months or more, to show significant improvement. In some cases, neurological damage may be permanent if treatment was delayed.
Can I drink alcohol if I have Wilson’s disease?
It is strongly recommended to avoid alcohol. Alcohol puts additional stress on the liver, which is already compromised by copper accumulation. Even small amounts can worsen liver damage and interfere with recovery.


Tallulah Sandison
May 4, 2026 AT 11:53omg this is so helpful thanks for posting!!
nikki paurillo
May 4, 2026 AT 22:52The metaphor of the clogged drain is strikingly visceral, isn't it? It paints a picture of internal stagnation that feels both mechanical and deeply organic. We often think of our bodies as fortresses, but they are really more like intricate ecosystems requiring constant flow. When that flow halts, the toxicity isn't just physical; it becomes a narrative of neglect written in copper deposits. I find myself pondering how we treat our internal landscapes with such disregard until the foundation cracks. The idea of ceruloplasmin acting as a guardian angel for transport is poetic in its biological precision. It reminds me of the old tales where spirits carry souls across rivers, preventing them from drowning in the mundane. Here, the protein carries metal, preventing us from drowning in our own chemistry. It is a delicate dance between accumulation and excretion, a balance that defines health as much as illness defines decay. Perhaps we should view every meal not just as fuel, but as a potential flood or a gentle rain. The liver, that silent gatekeeper, deserves more credit than it gets in our daily rush. It works tirelessly to filter the world's bounty into safe passage. When it fails, the consequences ripple outwards, touching the mind and the spirit. This disease teaches us about the fragility of homeostasis, that golden mean we strive for but rarely achieve. It is a reminder that even essential elements can become enemies when their rhythm is disrupted.
Ken Baldridge
May 6, 2026 AT 00:33Yo, let's break down the pathophysiology here because the ATP7B gene mutation is basically a system failure in the hepatic copper trafficking protocol. We're looking at a classic autosomal recessive defect where the transmembrane P-type ATPase enzyme loses its ability to load copper onto apoceruloplasmin or shuttle it into the biliary canalicular network. This results in progressive copper overload in the hepatocytes, leading to oxidative stress via Fenton reactions and subsequent cellular necrosis. The clinical presentation is bifurcated into hepatic and neuropsychiatric phenotypes, which is why early intervention is critical for preserving basal ganglia integrity. D-penicillamine acts as a potent chelating agent by forming water-soluble complexes with copper, facilitating renal excretion, but you gotta watch out for the transient neurological exacerbation due to rapid mobilization of brain copper. Trientine is a safer alternative for neuro-Wilson's patients because it has a lower affinity for brain copper, reducing the risk of worsening symptoms. Zinc acetate induces metallothionein synthesis in enterocytes, which binds dietary copper and prevents its absorption, making it ideal for maintenance therapy. The key takeaway is that adherence to lifelong pharmacotherapy is non-negotiable for maintaining biochemical stability and preventing irreversible organ damage. Regular monitoring of 24-hour urinary copper excretion and serum free copper levels is essential for titrating therapy effectively.
Bradley Gusick
May 6, 2026 AT 16:29You think this is just a genetic fluke? Think again. The pharmaceutical industry profits off chronic diseases, and Wilson's is no exception. They want you on penicillamine for life, creating a steady stream of revenue while ignoring the root causes. Copper accumulation is linked to environmental toxins and poor diet pushed by Big Food. The government knows this but stays silent to protect corporate interests. Wake up! Your body is under attack from multiple fronts, and these 'treatments' are just band-aids on a bullet wound. The real cure is detoxification through natural means, not synthetic drugs that poison your kidneys. Don't let them tell you it's all in your genes. It's in your environment, controlled by those who benefit from your sickness. Question everything. Demand transparency. The truth is out there, hidden behind layers of medical jargon and profit margins.
Robert Cowley
May 7, 2026 AT 03:04Oh, please. Another article trying to simplify complex biochemistry into cute metaphors. 'Imagine your body is a house.' Really? That's the best analogy? It reeks of oversimplification designed to placate the masses. The reality is far grimmer and less poetic. Copper toxicity doesn't just 'back up'; it catalyzes free radical formation, leading to lipid peroxidation and mitochondrial dysfunction. And don't get me started on the 'good news' part. Managing Wilson's isn't living a normal life; it's living a life dictated by pill schedules and blood tests. The author glosses over the severe side effects of chelation therapy, like anaphylaxis and bone marrow suppression. It's dangerous to paint such a rosy picture. People need to know the harsh truth: this is a debilitating condition that requires relentless vigilance. No amount of positive thinking will stop the copper from destroying your liver. Stop sugarcoating it.
Rebekah Korak
May 7, 2026 AT 22:44Let us delve deeper into the philosophical implications of copper accumulation, shall we? For what is Wilson's disease if not a manifestation of the universe's inherent imbalance? We consume, we accumulate, we fail to expel. Is this not the human condition? We hoard knowledge, wealth, and emotions, only to find ourselves poisoned by our own excesses. The liver, that ancient seat of anger and passion, becomes a tomb for unprocessed metals. The Kayser-Fleischer rings are not merely diagnostic markers; they are windows into the soul, revealing the golden burden we carry. To diagnose is to judge, to label, to categorize. But who are we to judge the ATP7B gene? It is simply following its programming, flawed as it may be. The chelation therapy is a form of purging, a ritualistic cleansing that mirrors ancient rites of passage. Yet, does it cleanse the spirit? Or merely the bloodstream? One must consider the existential weight of carrying a toxic legacy. Are we defined by our defects, or by our ability to manage them? The answer lies not in the medicine, but in the mindset. Embrace the copper. Let it teach you about limits, about boundaries, about the necessity of release. Only then can one truly transcend the physical constraints of the disease.
Lando Neal
May 8, 2026 AT 01:31I've been researching this for my cousin who was recently diagnosed, and I have to say, the clarity of this explanation is refreshing;; it cuts through the usual medical jargon;; I appreciate how you highlighted the difference between liver and neurological symptoms;; it makes sense now why some people present differently;; the part about zinc blocking absorption is fascinating;; I never realized the gut played such a direct role in trapping copper;; it's almost like a biological trapdoor;; I'm curious if anyone else has noticed changes in mood before the physical symptoms appeared;; my cousin mentioned feeling unusually anxious months before his tremors started;; it would be great to hear more personal experiences regarding the emotional toll;; knowing what to expect mentally could be just as important as understanding the physical treatment;; thanks for sharing this detailed breakdown;; it gives me hope that proper management is possible;;
Srinivas Komakula
May 9, 2026 AT 08:22The pathomechanism described herein is consistent with current literature regarding ATP7B mutations;; however, one must scrutinize the efficacy of standard chelation protocols;; recent studies suggest that tetrathiomolybdate may offer superior neuroprotection by inhibiting copper absorption and binding plasma copper without disrupting intracellular copper stores;; this distinction is critical in preventing the paradoxical worsening of neurological symptoms often observed with D-penicillamine initiation;; furthermore, the role of oxidative stress in neuronal degeneration cannot be overstated;; thus, adjunctive antioxidant therapy might be warranted in specific clinical scenarios;; the diagnostic criteria outlined are robust, yet genetic testing remains the gold standard for definitive confirmation;; it is imperative to emphasize the importance of family screening to identify asymptomatic carriers early;; proactive management can prevent irreversible hepatic or neurological damage;; therefore, a multidisciplinary approach involving hepatologists, neurologists, and genetic counselors is essential;;
Preety Singh
May 9, 2026 AT 18:05It is quite amusing how laypeople attempt to grasp complex metabolic disorders through simplistic analogies;; the notion of 'clogged drains' is intellectually lazy;; Wilson's disease is a sophisticated disorder of copper homeostasis governed by intricate genetic pathways;; those who suffer from it require precise medical intervention not folk wisdom;; the mention of diet restrictions is adequate but lacks nuance;; avoiding shellfish is trivial compared to the rigorous pharmacological regimen required;; one must adhere strictly to chelation therapy or zinc supplementation;; deviation leads to catastrophic outcomes;; do not mistake this for a lifestyle choice;; it is a life-threatening condition demanding expert management;;
Leah Sentz
May 9, 2026 AT 23:38This is so scary 😱 My friend has something similar and she always complains about her meds 💊 I hate seeing people suffer like that 😢 The eye rings thing is creepy 👀 Why does copper go to the eyes?! 🤔 I hope she gets better soon ❤️ Doctors are weird sometimes 🙄 But at least there is treatment right? 🤞
Sarah Mifsud
May 11, 2026 AT 14:29Great info! Just a small note tho, some people might confuse zinc with other supplements so make sure to specify zinc acetate specifically as it has better bioavailability for this purpose. Also, taking zinc with food can reduce nausea but might slightly decrease absorption so its a tradeoff. Hope this helps anyone reading!
Christina Lancey
May 13, 2026 AT 12:40It is wonderful to see such comprehensive information shared openly. Understanding these conditions reduces stigma and fear. Remember that every patient's journey is unique, and patience is key during treatment adjustments. You are not alone in this.